
Cystic Fibrosis Research News № 6
Inserting More Mutations May Improve CFTR Function
Our understanding of the underlying biology of cystic fibrosis has
grown more sophisticated over several decades: from the
determination in 1989
that CF is driven by the protein CFTR to our current capacity to
model the protein’s structure with computer simulations.
During this time, innovative new therapies have grown in tandem
with our biological understanding. The suite of drugs to treat the
underlying cause of CF already contains FDA-approved CFTR
modulators, and development is underway for gene therapy, mRNA
therapy, and other next-generation drugs.
Researchers in the UK and Canada are developing an even more novel
and somewhat curious therapeutic approach – mutating CFTR in a
second location to compensate for the problems caused by its
initial mutation. Early data is very promising, and new
therapeutic approaches that come out of this research may
complement current modulator drugs and even offer new strategies
for gene therapy.
One would not expect that intentionally adding mutations to an
already-defective gene would improve its function. Yet from a
molecular perspective, this approach may be helpful. The CFTR
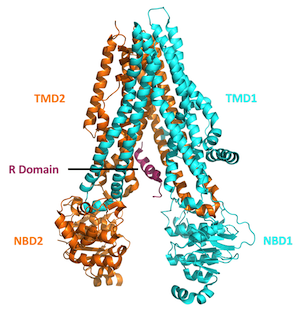
protein is made up of four domains or regions: two nucleotide
binding domains that are situated within the cell interior plus
two transmembrane domains that span the outer membrane of the cell
and form the pore of the CFTR protein channel. The Delta-F508
mutation (the most common among CF patients) is located within one
of the two nuclear binding domains.
A protein is essentially a long string of molecules called amino acids, folded up in a very precise orientation. For people with the Delta-F508 mutation, the amino acid phenylalanine, which would normally occupy the 508th position in this string of amino acids, is deleted. This leads to protein instability, and early degradation by the cell.
Why does the deletion of one amino acid cause the whole protein to become unstable? The amino acid phenylalanine contains an aromatic (ring-like) structure which engages in 'hydrophobic’, stabilizing interactions with other similar amino acids in the immediate vicinity. The deletion of F508 diminishes these interactions and thus the protein tends to fall apart. For the same reason, a well-placed mutation inserted into the same region of the protein may re-introduce stability by adding new chemical structures that are capable of engaging in hydrophobic interactions.
This is exactly what the researchers achieved – they used a technique called site-directed mutagenesis to introduce a variety of new mutations into one of the nuclear binding domains of CFTR and see if these mutations could compensate for the dysfunction caused by Delta-F508. Ultimately, they found one mutation - labeled R1070W because it replaces the amino acid arginine (R) at position 1070 in the string of amino acids with another amino acid called tryptophan (W) - that did the job quite well. When the CFTR protein containing this R1070W mutation was assessed for its ability to conduct ions across the cell membrane (as functional CFTR should), it performed far better than Delta-F508 mutant CFTR and reasonably close to normal CFTR.
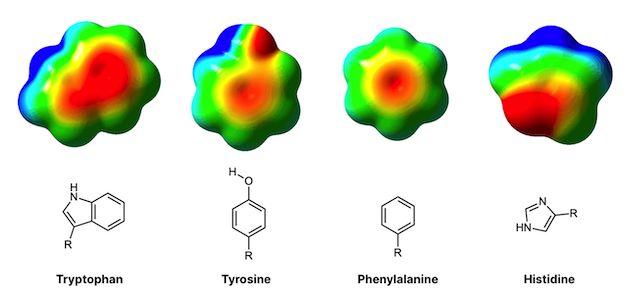
Why is this manually-inserted mutation so effective? The study employed sophisticated, computer-based molecular dynamics simulations, which showed that tryptophane possesses an aromatic group just like phenylalanine, whereas arginine (the amino acid typically found at position 1070), does not have an aromatic functional group. The researchers believe that introducing tryptophan compensates for the loss of phenylalanine because the two amino acids are similar in structure. Tryptophan re-introduces some of the chemical stability that the missing phenylalanine helped provide. Although F508 and W1070 are found at distant positions on the string of amino acids (562 amino acids apart, to be exact), proteins like CFTR are so intricately folded that the amino acids far apart in sequence actually may be found very close together in physical space (as is the case here).
This study has very interesting translational implications. For one, if the R1070W mutation does increase CFTR stability, this mutational approach may be used in conjunction with CFTR modulators to make a combined therapy that is even more effective. Furthermore, the mutational approach may also provide a potentially more feasible strategy for CF gene therapy. Instead of attempting to replace the mutant version of the CFTR gene with a functional copy, which involves a complicated cellular process called homology-directed repair (HDR) and has shown limited success in the past, researchers may instead focus on introducing new mutations using gene editing techniques, which is mechanistically more simple and may be more viable as a therapeutic strategy.
Featured Article: Prins S, Corradi V, Sheppard DN, Tieleman DP, Vergani P Can two wrongs make a right? F508del-CFTR ion channel rescue by second-site mutations in its transmembrane domains. J Biol Chem.2022;101615. doi:10.1016/j.jbc.2022.101615.
Computational Drug Design For New Antibiotics Against Pseudomonas
For the many people with CF colonized by the common pathogen
Pseudomonas aeruginosa, new therapies that can neutralize the
negative effects of the bacteria are much needed. Existing
antibiotic therapies like Tobramycin help stabilize lung function,
but bacteria tend to persist in the lungs, leaving the door open
for further therapies to target the bacteria and perhaps improve
pulmonary function even further. One research team from Concordia
University in Wisconsin is pursuing a highly targeted molecular
approach for developing new antibiotics.
The team is focused on a Pseudomonas protein called ExoU, which is
a virulence factor secreted by Pseudomonas that has the capacity
to infiltrate lung cells and chew up the cell membrane. This
contributes to cell death, and on a broader level, lung tissue
damage. The goal of this study was to develop a protocol for
screening novel drug candidates that could bind to ExoU and
prevent it from contributing to lung damage.
A drug screening protocol was devised that combined computational
and wet-bench techniques: computer modeling allowed the
researchers to identify potential sites on the ExoU protein where
a drug called methoctramine was likely to bind, and the wet-bench
experiments confirmed if the drug did indeed bind to the protein
when the two were combined in solution. The researchers chose the
drug methoctramine in particular because it has been shown in
animal studies to have a
broad range of clinical effects
such as, managing bradycardia, or abnormally slow heart rate, and
reducing memory impairment. Furthermore, the researchers had
already conducted previous research which suggested that
methoctramine may impair ExoU function.
Today’s computer software for drug design is incredibly powerful,
and sophisticated enough to enable the researchers to identify a
top-ranked drug binding site and pinpoint its exact location to a
region on the C-terminal end of the ExoU protein.
Computational drug design takes into account factors like the
orientation of a protein’s amino acids in 3-Dimensional space, as
well as the chemical characteristics of individual amino acids in
particular regions of the protein. A binding site is deemed high
quality if the atoms of a drug are likely to interact strongly
with the amino acids of the site. That said, other factors such as
the hydrophobicity of the amino acids in the binding site can
negatively affect its quality even if it has a good chemical
affinity for the drug. This is because the amino acids that are
more hydrophobic have a lower affinity for water (cells are
essentially bags of water that proteins floating around in) and
thus tend to bunch up in the center of a protein, largely
inaccessible to therapeutic compounds.
After computational analysis determined that methoctramine was
likely to interact with ExoU, the researchers relied on a
laboratory assay called nucleic magnetic resonance (NMR) to prove
that methoctramine does bind to ExoU in the real world (and not
just in the computer simulation). The NMR results indicated that
the two molecules did indeed interact, and permitted an estimation
of the dissociation constant (KD), a measure of how tightly two
molecules are bound. The research noted that the KD value,
estimated at 1.3 x 10-5 M/ L, was well within the range of values
considered to be strong binding for this sort of experiment.
Going forward, the Concordia researchers believe that
methoctramine might be chemically modified to bind even more
tightly to ExoU, and tested in animal models of Pseudomonas lung
infection to demonstrate clinical efficacy as a drug, and further
modified to minimize toxicity and maximize pharmacokinetic
parameters of absorption and distribution throughout the body. If
effective at reducing bacterial virulence, this approach may help
CF patients recently colonized with Pseudomonas. And although
chronic infection isolates tend to express ExoU less
significantly, this type of of combined computational / wet-bench
approach may be applied to the testing and discovery of other
molecules to target Pseudomonas.
Featured Article: Chamberlain K, Johnson M, Reid TE, Springer TI. Utilizing in silico and in vitro methods to identify possible binding sites of a novel ligand against Pseudomonas aeruginosa phospholipase toxin ExoU. Biochem Biophys Rep. 2021;29:101188. Published 2021 Dec 16. doi:10.1016/j.bbrep.2021.101188.
Therapeutic Approaches May Improve CF Bone Health
In recent years, researchers have begun to focus more intently on
the extra-pulmonary symptoms of CF that tend to present over time,
but may have been overshadowed by the specter of lung disease in
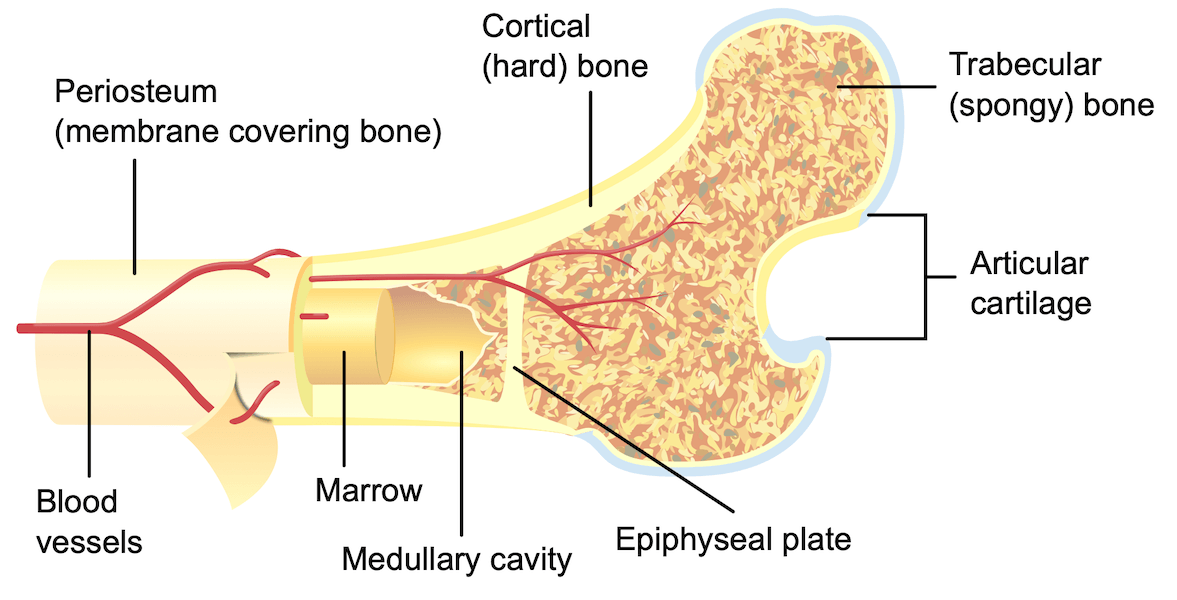
the past. One such area of focus is CF bone disease. It is well
established that the bones of people with CF do not grow and heal
as well, leaving CF patients more prone to fracture, and more
likely to experience excessively long periods of healing after a
bone break. A team of researchers from several Chinese
universities, is developing and testing bone implants that can
overcome both underlying bone weakness and delayed bone healing.
With careful description, the researchers note that CF bone
disease has many biological causes. Among them are procollagen 1
N-terminal peptide (PINP). This molecular marker of bone formation
was found to be less prevalent in the serum of Delta-F508 mutant
CF mice.
The study showed that the Wnt/Beta-catenin signaling pathway,
which is crucial for bone formation, is also down-regulated. And
the Mineral apposition rate (MAR) – a measure of how long it takes
for new bone to be produced after a fracture – is diminished.
Studies have suggested underlying causes for CF bone disease, from
reduced calcium uptake due to nutrient malabsorption in the gut,
to defects in the CFTR protein in bone cells themselves
influencing bone growth. Bone disease is likely a combination of
multiple such factors.
Fortunately, new treatment approaches seem able to compensate for
CF-related bone defects. The study combined engineering techniques
with biochemical approaches to develop a magnesium implant (a
metal rod inserted into the femur) that allowed Delta-F508 mutant
CF mice to regrow bone at rates similar to mice without CF.
To conduct their experiments, the researchers broke the bones of
an unfortunate group of mice (both CF and non-CF), taking care to
anesthetize them first, then went to work on fixing the fractures.
The CF mice treated with standard implants (stainless steel, not
magnesium) demonstrated less rapid bone growth, and their levels
of the bone formation marker PINP were also decreased. However,
when the CF mice were given the magnesium implants, they
demonstrated bone growth that more closely matched that of non-CF
mice.
This finding is certainly exciting for people with CF who happen
to suffer a bone break. But what about the larger group of people
with CF who have not suffered a bone break yet, but who have
presented with lower bone density at the clinic, and therefore are
at higher risk for fracture. In addition to testing magnesium
implants, the research team also tested the effects of magnesium
supplements on CF mice. It was revealed that oral magnesium
supplements improve bone health by a number of measures, including
bone density in DXA scans, PINP levels and Wnt/Beta Catenin
pathway activation. While this effect is not as strong as that of
the implants, it is an indication that magnesium supplementation
should be studied and further considered when contemplating
standard CF care.
Featured Article: Xu J, Hu P, Zhang X, et al. Magnesium implantation or supplementation ameliorates bone disorder in CFTR-mutant mice through an ATF4-dependent Wnt/β-catenin signaling. Bioact Mater. 2021;8:95-108. Published 2021 Jul 3. doi:10.1016/j.bioactmat.2021.06.034.
Featured Five CF Stories
It’s impossible to list all of the amazing research that is
on-going for CF. Below is a quick list of a few fascinating
articles that seem to show significant promise.
One Mutant CFTR Allele Worsens Effects of Smoking: Observational study reveals that cigarette smokers who possess just one mutant CFTR allele are more likely to develop bronchitis. This is in line with the findings of other studies that heterozygotes are not free of clinical symptoms – rather, they are likely to possess a more mild form of the clinical symptoms of CF that may be less noticeable or present only in certain circumstances, like exposure to cigarette smoke. (European Respiratory Journal).
New Nutritional Measure to Predict CF Clinical Outcomes: Cross-Sectional study finds a positive correlation between lung function and lean body mass. Furthermore, higher levels of physical activity were associated with increased lean body mass in this study – suggesting that it may help boost lung function. The authors suggest that lean body mass, particularly a measure called appendicular lean mass index (ALMI), might be a better predictor of lung function than BMI, the traditional metric used to assess nutritional health in CF patients. (Nutrients).
Improved Lung Transplantation Among CF Patients in France: A review of the High Emergency Lung Transplantation (HELT) program in France revealed that it significantly boosted the number of transplantations in the period after its implementation. It also ensured that the number of CF patients in France who received a transplant between 2008 and 2016 was significantly higher than the number of patients who died waiting. The authors suggest that the French program should be emulated in other countries as well to save lives. (European Respiratory Journal).
CF From a Global Perspective: The latest data on CF across the world estimates that 162,428 people are living with CF (at least in countries where a patient registry exists and total population is greater than 1 million people). While regulatory approval for CF modulators continues to expand to more countries, the study estimates that only 12% of people with CF worldwide are receiving triple combination drugs so far. Furthermore, it is estimated that only 65% of CF patients across the world have actually been diagnosed with the disease – spotlighting the need for efforts to make CF screening measures more effective and accessible in certain countries. (Journal of Cystic Fibrosis).
Probiotics and Prebiotics Might Help Treat Stubborn GI Symptoms: Recent review weighs the evidence in favor of probiotic and prebiotic use as a way to resolve inflammation in the GI tract of CF patients. Early evidence from randomized control trials and epidemiological studies support the use of both measures as a means to reduce gut inflammation in general, though the evidence of their effectiveness in a CF-specific context is still rather limited. As lung health has dramatically improved following the approval of CF modulators, clinicians and researchers are focusing more on the gut microbiome and gut-related symptoms (as well as other non-pulmonary aspects of CF) as key unresolved components of the disease. (Nutrients).
Clinical Trial Watch
The latest news on CF drug development and clinical trials.
International, 17 Sites. Recruiting: New Vertex trial is further assessing the positive impact of CF modulator therapy, beyond measuring FEV1 (lung function) as a primary outcome and looking at more general physiological symptoms: percent reduction in cough frequency from baseline and change in baseline total step count per day (before and after starting therapy). Both measures will be tracked with wearable technology. (Vertex Pharmaceuticals).
Atlanta, Georgia. Recruiting: The gold standard for CF diagnosis is the sweat test. It requires that the person being tested wear a small cuff-like device around their arm for roughly half an hour that uses electric current to induce sweating. Sometimes, the procedure has to be performed multiple times before a patient produces enough sweat to make an accurate diagnosis. The researchers conducting this study will test two alternative methods on healthy individuals without CF – a pilocarpine microneedle patch, and pilocarpine iontophoresis (which uses a gel disc to deliver medicine) to see if they (a) generate more sweat than the traditional sweat test, minimizing need for multiple tests, and (b) measure sweat chloride concentration effectively. (Emory University).
Dallas, Texas. Recruiting: Given that bone disease is a common co-morbidity for CF patients, researchers want to develop a better understanding of bone disease markers. The goal is to gather 100 subjects – with and without CF – to participate in a single study visit and undergo a DEXA bone scan, a Micro CT scan, and blood tests. Individuals who show signs of bone disease at this visit will be recommended for Denosomab treatment (a monoclonal antibody therapy) and asked to complete annual follow-up visits (performing the same tests) for the next five years. (University of Texas Southwestern Medical Center).
British Columbia, Canada. Recruiting: Many in the CF research community are eager to understand long-term patient health trends following the initiation of CF modulator therapy. This Canadian study will gather a variety of data on pulmonary symptoms, nutritional effects, quality of life scores, physical activity, and more. The stufy also will build a biorepository of clinical samples from modulator-treated patients that will provide valuable experimental data for years to come. (University of British Columbia).
Funding News
Recent funding for CF researchers or companies.
Funding for New antibiotics and Better Drug Delivery: The Cystic Fibrosis Foundation (CFF) continues to provide strong funding to a variety of companies pursuing novel CF therapies. The foundation awarded $3.5 million to Aceragen to assist their development of an antibiotic to treat drug-resistant S. aureus bacteria. The FDA has already granted Aceragen an Orphan Drug designation for the antibiotic. CFF is also supporting Feldan Therapeutics with money out of its $500 million Path to a Cure initiative. The company is developing their patented ‘Feldan Shuttle’ technology to delivery medicines more effectively to the epithelial cells of the lungs. (Aceragen Inc., Feldan Therapeutics).
CF Pathogen Research Gets Big Boost from the CFF: CFF reports that it has contributed funds exceeding $109 million dollars over the past three years towards the Infection Research Initiative, with the goal of improving treatment and detection of chronic infections. From its inception, the initiative has funded 20+ industry programs and 200+ academic labs in the pursuit of better diagnostic tests, more effective antibiotic regimens, and a fuller understanding of how CF infections are impacted by modulator therapy. (Cystic Fibrosis Foundation).
Better Pancreatic Enzymes are on the Way: The company First Wave Biopharma has filed two new patents for their yeast-derived enzyme (adrulipase) to fight pancreatic insufficiency (PI) in CF. Clinical trials have shown that combining adrulipase with standard pancreatic enzyme replacement therapy (pig-derived enzymes) has the potential to further improve the digestion of fats from food. A more effective PERT regimen would stand to improve nutrition and resolve GI symptoms for people with CF. (First Wave Biopharma).
Expanding Options for People with Rare CFTR Mutations: The EU-Funded HIT-CF program is finding success in their efforts to test CFTR modulators on organoids (lab-grown, miniature organ-like structures) from patients with ultra-rare CFTR mutations. Ultra-rare mutations are so rare that only 1-2 people in the world possess them. It is estimated that roughly 5,000 people in Europe fall into this category. Thus far, the program has grown organoids from 500 different patients, and is collaborating with pharmaceutical companies with plans to test drug compounds on the organoids starting in April. (Horizon: The EU Research & Innovation Magazine).
A Call to Action
Cystic fibrosis (CF) research is very much dependent on the
strength of the CF community. It’s not simply an effort carried
out by scientists in white lab coats - although there are many of
them, and their work has enormous impact. Advances in research
also depend on the technicians and engineers who operate the
laboratory equipment that enables drug discovery, and the
industrial machinery that allows drug development. Research
depends on both business and marketing professionals, those who
make biopharma companies viable and promote clinical trials.
Successful research further depends on clinical trial
coordinators, who carry out studies and work tirelessly to recruit
and support patients throughout the complicated trial process.
Particularly for rare diseases like cystic fibrosis, research
depends on the work of foundations and patient advocates, which
includes in the United States organizations such as the CF
Foundation, Emily’s Entourage, CFRI, and the Boomer Esiason
Foundation, as well as countless other across the globe, and
hundreds of committed clinicians and researchers. Most
importantly, research depends on people with CF and their devoted
families and friends.
There can be no progress in CF research without patients willing
to participate in clinical trials: not only to test new drugs, but
also to provide, quite literally, their flesh and blood. It is
with the help of patient samples that scientists can understand
the damage that CF inflicts upon the human body, and also how
drugs developed by the research community can remedy these
damages.
This newsletter aims to pull all of these threads together;
allowing the CF community to more fully appreciate how well the
aims of its many members are aligned (and it extends an invitation
to all readers not yet a part of the CF community, to embrace the
cause and take up the task of pushing CF research forward).
There’s something here for everyone - those interested in the
clinical side of CF care, or in drug development, or the technical
work performed in CF-centered laboratories. The newsletter also
has as its objective to showcase new clinical trials; an
opportunity for patients and clinicians to take part. Wherever and
whoever you are in the world, you too may push CF research forward
- either by direct participation, or simply by reading and sharing
this newsletter with others.